Front End Innovation
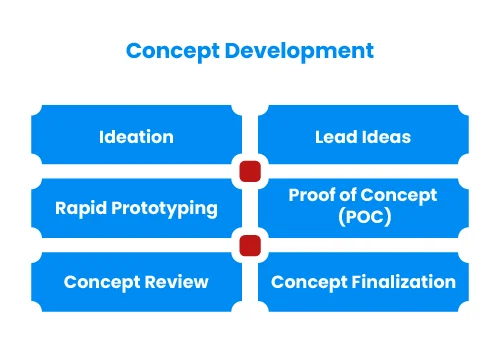
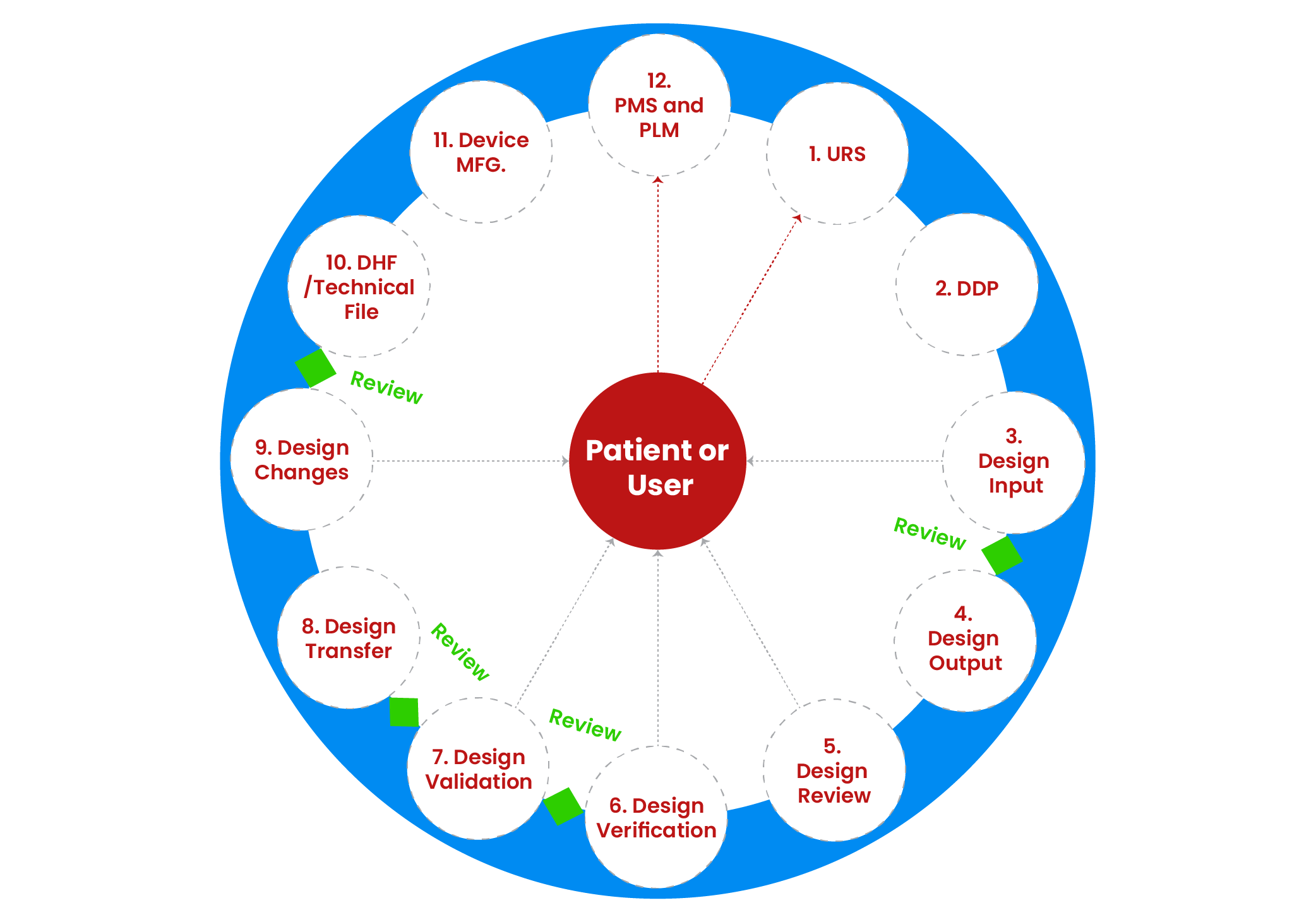
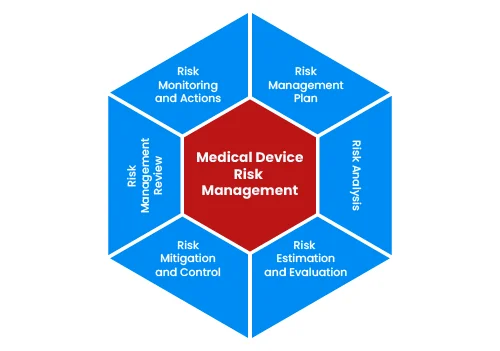
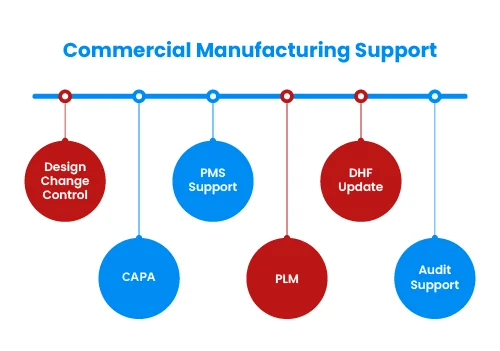
Medical device development starts with identifying unmet clinical need or business opportunity. This can be done through various methodologies such as User Research, Voice Of Customer (VOC), Job Outcome Constraints (JOC) or contextual innovation. We will help you in questionnaire design, conduct qualitative market research to collect primary data and report findings. This will result into defining User Requirement Specifications (URS) or need criteria at broader level.